Biography: I am a pharmacy student at University College London with a strong interest in…

Researcher Spotlight – Lucy Williams – Runner-up of the BSGCT Scientific Writing Competition
Gene-ious at Work: Editing the Future for Patients with Sickle Cell Disease
By Lucy Williams, PhD student at Cardiff University and runner-up of the BSGCT Scientific Writing Competition.
Lucy’s research focuses on the development of a precise virus-based cancer therapy that overcomes the limitations of existing treatments by specifically targeting cancerous cells and evading patients’ anti-viral immunity. Another strand of her work aims to develop a viral vector capable of being deployed as a ‘cancer vaccine’ or adapted for the delivery of gene therapy.
Picture a red blood cell (RBC). A scarlet disc, floating along the highway of our bloodstream. Their signature colour owed to the iron contained in their haemoglobin (Hb). Taking on the role of a delivery van, the RBC uses Hb to transport oxygen from the lungs to our cells, then returns with the carbon dioxide waste which we breathe out. Indentations on either side of the disc’s surface create its characteristic biconcave shape, like a ball of plasticine squashed between a child’s little fingers. A flexible doughnut without a hole, RBCs can squeeze through the narrowest side-streets (capillaries) on their journey, and their large surface area maximises the oxygen they can carry. The Hb used to haul oxygen around the body comes in a few different varieties, with the key ones in healthy individuals being foetal haemoglobin (HbF) and adult haemoglobin (HbA). Your body makes HbF when you are inside your mother’s womb and is made up of proteins called α-globin and γ-globin. Shortly after birth, your body reduces its production of HbF and starts making HbA composed of α-globin and β-globin proteins instead [1].
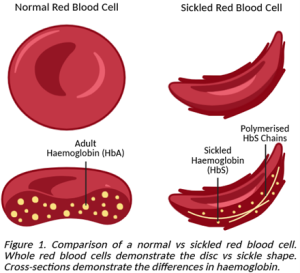
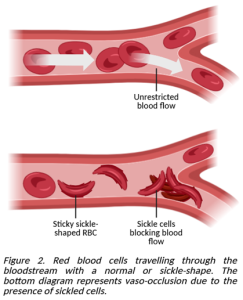
Sickle cell disease (SCD) refers to a group of inherited conditions that impact how RBCs develop and function, principally in those of African or Caribbean descent. A mutation in the gene responsible for directing RBCs to make β-globin results in the production of abnormal sickle haemoglobin (HbS), which derives its name from the corn cutting farmer’s tool that an RBC with HbS resembles. HbS is sticky and prone to clumping together into rigid chains that pull the RBC into a stiff crescent, making it difficult for them to pass through capillaries as they attempt to move around the body. Getting stuck – especially at width-restricted junctions – leads to a vaso-occlusive crisis (VOC) where the sickled cells block the flow of traffic in the blood, causing oftentimes severe pain in the back, chest, and limbs, as well as impairing downstream organ function. Some individuals with SCD also have a genetic disorder known as β-thalassemia, in which their RBCs can’t make enough of the adult form of haemoglobin (HbA), meaning that they aren’t as efficient at transporting oxygen [2].
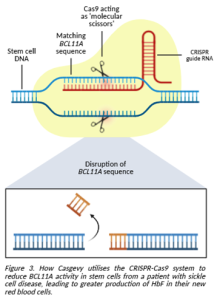
In 2023, a gene editing therapy called Casgevy was approved in the UK for treating SCD and β-thalassemia in patients who suffer from repeated VOCs and are eligible for a bone marrow transplant but have no appropriate donor available. Patients have stem cells collected from their bone marrow so that their genes can be edited in the lab using CRISPR-Cas9 technology. CRISPR is a short piece of RNA that acts as a set of instructions to guide the Cas9 enzyme towards a specific gene. Cas9 can then be used like a pair of molecular scissors, cutting the gene so that researchers can make changes using the cell’s natural repair mechanism. A custom DNA sequence works like a blueprint and can be added into the cell, allowing the machinery to repair the cut using this new sequence as a building block, just like fixing a mistake in a document using the “find and replace” function [3].
For treating SCD and β-thalassemia, the CRISPR-Cas9 system used in Casgevy targets a gene called BCL11A. This gene is responsible for telling the body to make less γ-globin for HbF and switch to producing more β-globin for HbA instead. Since it is the β-globin in HbA which is faulty, using gene editing to reduce the activity of BCL11A would promote the stem cell to turn into an RBC producing functional HbF with γ-globin instead [4].
While waiting for their stem cells to be edited, the patient must undergo chemotherapy to prepare their bone marrow by killing any remaining stem cells that would otherwise continue to produce faulty RBCs. This conditioning regimen also temporarily suppresses the patient’s immune system, minimising the risk of the newly transplanted cells being attacked and rejected by the body. Once edited, the stem cells are then infused back into the patient and given time to settle into their now spacious home in the bone marrow [5]. This increases the level of RBCs that don’t form a sickle shape, making them less likely to stick together and cause a VOC in the future. Editing stem cells rather than the RBCs themselves makes this therapy a one-time treatment designed to last the life of the stem cells. Although the longevity of transplanted cells depends on a variety of factors, their ability to self-renew means that they can survive for many years, or even a patient’s entire life [6].
A clinical trial investigating the use of Casgevy was initiated in 2018. When patients were evaluated 2 years post-treatment, over 90% had gone without a VOC for at least 12 consecutive months. Of those, only one had required hospitalisation in over 22 months and none of the patients experienced transplant rejection or failure [7]. Monitoring of these patients will continue in the hopes that this one-shot gene therapy provides lifelong protection against VOCs for those with SCD and β-thalassemia.
References
- Sankaran VG, Orkin SH. The Switch from Fetal to Adult Hemoglobin. Cold Spring Harbor Perspectives in Medicine [Internet]. 2012 Dec 3;3(1):a011643. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC3530042/
- Sickle cell crisis [Internet]. PubMed. 2025. Available from: https://pubmed.ncbi.nlm.nih.gov/30252320/
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols [Internet]. 2013 Oct 24;8(11):2281–308. Available from: https://www.nature.com/articles/nprot.2013.143
- How CASGEVY works [Internet]. CASGEVY® (Exagamglogene Autotemcel) Patient Website. Available from: https://www.casgevy.com/sickle-cell-disease/how-casgevy-works
- Maqbool S, Baloch MF, Khan MAK, Khalid A, Naimat K. Autologous hematopoietic stem cell transplantation conditioning regimens and chimeric antigen receptor T cell therapy in various diseases. World Journal of Transplantation [Internet]. 2024 Mar 15;14(1). Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC10989471/
- Chapman MS, Wilk CM, Boettcher S, Mitchell E, Dawson K, Williams N, et al. Clonal dynamics after allogeneic haematopoietic cell transplantation. Nature [Internet]. 2024 Oct 30;635(8040):926–34. Available from: https://www.nature.com/articles/s41586-024-08128-y
- Parums DV. Editorial: First regulatory approvals for CRISPR-CAS9 Therapeutic gene editing for sickle cell disease and Transfusion-Dependent Β-Thalassemia. Medical Science Monitor [Internet]. 2024 Feb 21;30. Available from: https://pubmed.ncbi.nlm.nih.gov/38425279/
Related Posts


