Gene-ious at Work: Editing the Future for Patients with Sickle Cell Disease By Lucy Williams, PhD…

A step in the right direction: A novel approach to gene therapy for Charcot-Marie-Tooth disease type 2
Author: Dr. Rebecca A Lea, University of Sheffield
Charcot-Marie-Tooth (CMT) disease is an umbrella term that encompasses a broad range of genetic neurodegenerative diseases, first described by doctors in 1886 [1, 2]. It is characterised by symptoms such as muscle weakness or paralysis starting in the feet and legs, balance problems, muscle cramping and nerve pain [3], all resulting from the death of specialised cells known as motor neurons, the nerve cells that control movement. As a whole, CMT affects approximately 1 in 2500 people, making it the most common form of inherited peripheral neuropathy [4], that is, a disease affecting the health of nerve cells that connect the brain and spinal cord (the central nervous system) to rest of the body (the periphery). To complicate matters, CMT is commonly divided into three distinct categories with multiple sub-types each, and mutations in more than 90 genes have been linked to the presence of disease [5]. Among the sub-types of disease, CMT type 2A (CMT2A) is one of the most common, accounting for up to about 5% of all diagnoses [6]. It is also one of the most severe sub-types, with symptoms often initiating at a young age and patients commonly losing the ability to walk independently by the age of 21 [7]. There is therefore a dire need for effective treatments for this devastating disease.
Currently, however, there is no curative treatment for CMT2A. Therapy mostly revolves around management of the disease and its symptoms, including for example physical therapy and walking aids to maintain the patients’ mobility and independence for as long as possible [8]. However, such interventions cannot target the cause of the disease and, as degeneration of the motor neurons continues over time, the patient’s condition inevitably worsens.
The cause of CMT2A is any one of a large number of mutations in a gene called Mitofusin 2 (MFN2), with more than 100 mutations identified to date [9]. The protein encoded by MFN2 has many roles associated with mitochondria, the energy-producing structures inside our cells, and mutations associated with CMT2A have been shown to lead to changes in mitochondrial size, shape and transport throughout motor neurons [10]. Nerve cells have an exceptionally high demand for energy, so these mitochondrial defects resulting from MFN2 mutations lead to the degeneration of motor neurons and the subsequent debilitating symptoms of CMT2A. However, the exact mechanism by which this occurs is still not understood, and this lack of understanding has hampered the development of drugs.
Since the causative gene is well-defined, one would imagine that CMT2A would be an excellent target for the use of gene therapy to “fix” mutant MFN2 and cure the disease. However, the huge variety of known CMT2A-causing mutations complicates matters, and while personalised medicine approaches targeting each patient’s specific mutation are technically feasible, they are both costly and time consuming and therefore likely to be inaccessible for most patients. As such, we need an effective, one-size-fits-all approach to tackle CMT2A at its cause.
This is where recent work from scientists in Milan comes in [11]. To target CMT2A, Rizzo and colleagues designed and tested a universal strategy to inhibit the production of all native MFN2 (nMFN2) protein in cells, while simultaneously providing a healthy copy of the gene to “rescue” normal expression levels. They call this approach “combined RNA interference and gene replacement therapy” or “RNAi/gene therapy” for short. RNA interference (RNAi) is a phenomenon whereby short molecules of RNA that are targeted to bind to specific gene sequences lead to a block of protein production from such genes, usually by encouraging the degradation of the intermediate gene products (see Figure 1 below) [12]. This block is not 100% effective, however, meaning that some native protein can still be produced. But in this new approach, it was hoped that the levels of native MFN2 would be sufficiently reduced that the “rescue” MFN2 (recombinant MFN2) protein would predominate. To protect rMFN2 from RNAi, Rizzo and colleagues engineered a number of changes to the RNAi-targeted MFN2 sequence (represented in magenta in Figure 1). These changes are “silent”, that is, they have no effect on the protein produced, but they prevent the short RNA from recognising rMFN2 and therefore enable the production of healthy MFN2 protein.
When tested in patient-derived motor neurons, the RNAi/gene therapy approach led to an efficient reduction of nMFN2 and restoration of protein expression via the introduction of rMFN2, proving that this combined approach could work in principle. Indeed, compared to untreated CMT2A patient motor neurons, RNAi/gene therapy-treated motor neurons showed an improvement of all mitochondrial defects. While this is a significant success at the cellular level, it is important to understand how the RNAi/gene therapy treatment would affect an entire organism. To this end, Rizzo and colleagues turned to an established mouse model of CMT2A, known as MitoCharc1, which expresses mutant MFN2 specifically in nerve cells [13]. The researchers were able to show the expected reduction of nMFN2 and expression of rMFN2 in mice exposed to an adenovirus harbouring the combined therapy. Unfortunately, however, even untreated MitoCharc1 mice did not show any significant signs of disease compared to healthy counterparts. It is a known issue, particularly in CMT, that many mouse models do not accurately recapitulate the progression and/or symptoms of human disease [14]. In this case, the MitoCharc1 mice, which were previously reported to exhibit relatively mild CMT2A-like symptoms [13], failed to develop such symptoms in the hands of the Milan researchers. Without this baseline measure of the effect of mutant MFN2 in the MitoCharc1 mice, it was impossible for Rizzo and colleagues to determine whether the rescue of healthy MFN2 expression was beneficial. Nonetheless, this experiment proved the potential of the RNAi/gene therapy approach to restore healthy expression of MFN2 and provided the first hint that such a treatment could be safe and effective for people with CMT2A.
Clearly, additional work would be needed to establish both the potential risks and potential benefits of the RNAi/gene therapy approach for CMT2A. Firstly, long-term safety and efficacy would have to be proven in animal models, the current gold-standard of testing for experimental therapies. Additionally, the sequence of the short RNA molecule for nMFN2-targeted RNAi would have to be chosen very carefully. The region to be targeted would have to be completely devoid of mutations, both disease-causing and benign, and also unique in the whole of human DNA, to prevent spurious targeting and dysregulation of other gene sequences.
Even so, the recent work by Rizzo and colleagues is a promising and exciting step towards a future treatment for people with CMT2A. Importantly, similar adenovirus-based gene therapies have a proven safety record, having been used already in more than 50 clinical trials of human gene therapies. And in a landmark success for the treatment of a neurodegenerative disease, an adenovirus-based gene therapy for spinal muscular atrophy has already been approved by the regulatory authorities in both the US and Europe (the FDA and EMA, respectively) [15-17]. With this in mind, and the evidence of molecular rescue in patient-derived cells, the combined RNAi/gene therapy approach for CMT2A, and indeed other genetic diseases, is certainly one to watch.
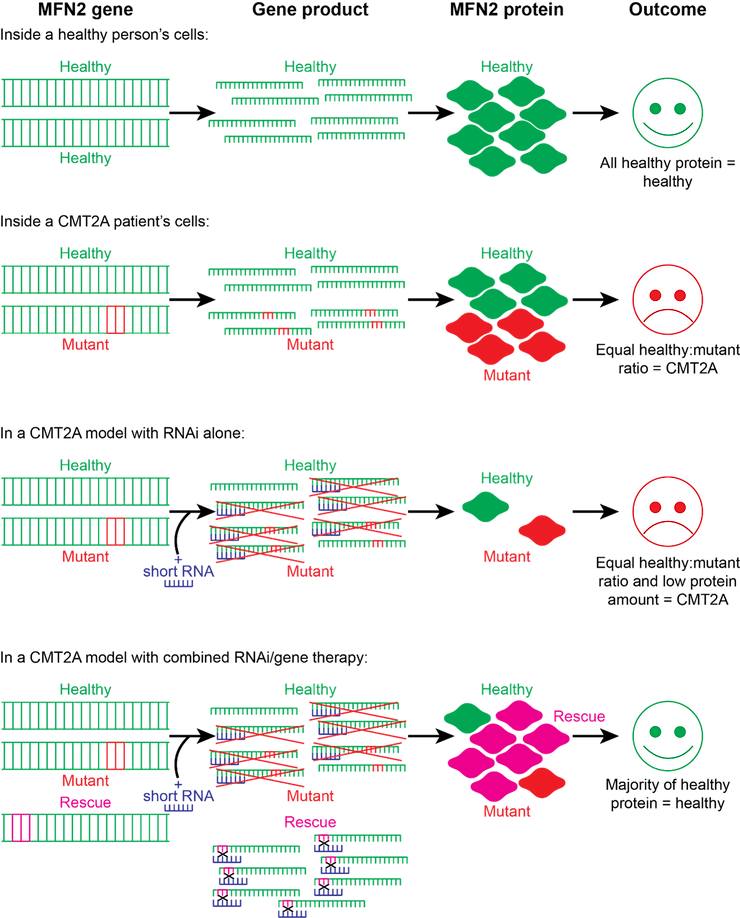
Figure 1: Illustrative summary of RNAi/gene therapy strategy for CMT2A. Most people carry two copies of the “healthy” MFN2 gene, leading to production of 100% healthy protein that is able to maintain mitochondrial and motor neuron health. CMT2A develops when a person has both a mutant copy of the MFN2 gene and a healthy copy. The mutant MFN2 protein overwhelms the healthy protein, causing the hallmark mitochondrial defects and motor neuron degeneration of CMT2A. The introduction of short RNA for RNAi leads to equal suppression of both healthy and mutant MFN2 genes (shown as red crosses) and is therefore unable to restore the healthy expression pattern. However, when a RNAi-resistant “rescue” copy of MFN2 is introduced into the DNA alongside the short RNA, the rescue protein is expressed in abundance, as the short RNA cannot bind (black crosses). This restores the normal MFN2 protein amount and allows healthy MFN2 (consisting of both the healthy native protein and the rescue protein) to outcompete the mutant version. With restoration of a normal expression pattern of healthy MFN2, mitochondrial and motor neuron health are maintained and symptoms are alleviated.
References:
1. Charcot, J.M., Sur une forme paticuliere d’atrophie musuculaire progressive souvent familial, debutante par les pieds et les jambes et atteignant plus tard les mains. Rev. Med Fr, 1886. 6: p. 97-138.
2. Tooth, H.H., The peroneal type of progressive muscular atrophy. 1886, University of Cambridge.
3. National Institute of Neuroligcal Disorders and Stroke, Charcot-Marie-Tooth Disease. Available from:
4. Skre, H., Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin Genet, 1974. 6(2): p. 98-118.
5. Pisciotta, C. and M.E. Shy, Neuropathy. Handb Clin Neurol, 2018. 148: p. 653-665.
6. Morena, J., A. Gupta, and J.C. Hoyle, Charcot-Marie-Tooth: From Molecules to Therapy. Int J Mol Sci, 2019. 20(14).
7. Feely, S.M., et al., MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology, 2011. 76(20): p. 1690-6.
8. National Health Service (NHS), Treatment: Charcot-Marie-Tooth disease. Available from: https://www.nhs.uk/conditions/charcot-marie-tooth-disease/treatment/.
9. Zuchner, S., et al., Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet, 2004. 36(5): p. 449-51.
10. Dorn, G.W., 2nd, Mitofusin 2 Dysfunction and Disease in Mice and Men. Front Physiol, 2020. 11: p. 782.
11. Rizzo, F., et al., Combined RNA interference and gene replacement therapy targeting MFN2 as proof of principle for the treatment of Charcot-Marie-Tooth type 2A. Cell Mol Life Sci, 2023. 80(12): p. 373.
12. Bagasra, O. and K.R. Prilliman, RNA interference: the molecular immune system. J Mol Histol, 2004. 35(6): p. 545-53.
13. Cartoni, R., et al., Expression of mitofusin 2(R94Q) in a transgenic mouse leads to Charcot-Marie-Tooth neuropathy type 2A. Brain, 2010. 133(Pt 5): p. 1460-9.
14. De Gioia, R., et al., Animal Models of CMT2A: State-of-art and Therapeutic Implications. Mol Neurobiol, 2020. 57(12): p. 5121-5129.
15. Mendell, J.R., et al., Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med, 2017. 377(18): p. 1713-1722.
16. U.S. Food & Drug Administration (FDA), FDA news release: FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. 2019; Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease#:~:text=The%20U.S.%20Food%20and%20Drug,genetic%20cause%20of%20infant%20mortality.
17. European Medicines Agency (EMA), New gene therapy to treat spinal muscular atrophy (corrected). 2020; Available from: https://www.ema.europa.eu/en/news/new-gene-therapy-treat-spinal-muscular-atrophy-corrected.
Related Posts


