Gene-ious at Work: Editing the Future for Patients with Sickle Cell Disease By Lucy Williams, PhD…

Researcher Spotlight – Ouidad Khechaoui – Runner-up of the BSGCT Scientific Writing Competition
After the Window Closes: The New Hope for Neurodevelopmental Disorders
By Ouidad Khechaoui, PhD researcher in neurodevelopmental AAV Gene Therapy at the University of Sheffield
When the parents of a child with a neurodevelopmental disorder first hear the diagnosis, they are often warned about a “critical window”, a short period in early childhood when treatment might make a difference. After that, the brain’s wiring was thought to be fixed, and any lost functions thought to be gone forever [1].
This window is frequently missed— not because families or clinicians fall short, but because symptoms can take years to appear, diagnoses are often delayed, and for many rare conditions, no treatments even exist during early development.
But that narrative is beginning to change. A new wave of gene therapies is challenging the long-held assumption that these disorders are irreversible after infancy. Emerging science shows that even when development goes off-course early, the brain retains a surprising capacity to respond, if the right genetic tools are delivered [2,3,4,5,6,7].
Challenging the Critical Window
For decades, researchers thought that the early absence or malfunction of genes essential to brain development, like MECP2, SCN1A, or SYNGAP1 [3,4,5], resulted in permanent neural damage. Medical experts used to agree that effective treatment needed to occur during the brain’s “plastic window” to be successful.
Yet preclinical studies have been quietly rewriting that rule. In mice with neurodevelopmental disorders, scientists have restored gene function in adulthood and seen remarkable results. Memory improved, seizures subsided, and lost behaviours re-emerged. The idea that the brain’s fate is sealed in infancy is no longer absolute.
A 2019 study on SYNGAP1-related intellectual disability serves as a key illustration of this advancement. The underexpression of this gene which controls synaptic plasticity—the brain’s ability to adapt and change— produces developmental delays leading to seizures and cognitive dysfunction. Researchers switched the SYNGAP1 gene back on in adult mice and found that seizure thresholds increased and long-term memory improved [3].
These findings proved that the adult brain could still respond to treatment, even after years without the gene. The window, it turned out, could reopen (Figure1)
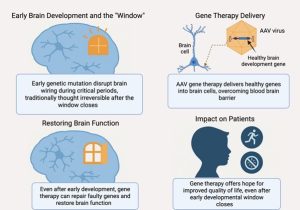
Figure 1: From Closed to Reopened – A Four-Panel Look at Gene Therapy’s Potential Beyond the Critical Window
Rett Syndrome: Reopening the Window
Perhaps the most significant clinical example comes from Rett syndrome, a rare and severe neurodevelopmental disorder caused by mutations in the MECP2 gene. Rett affects primarily girls and leads to profound intellectual disability, loss of speech, motor dysfunction, and seizures. Girls with Rett syndrome show typical development during their first two years before they start losing the new abilities they have recently acquired.
For years, that regression was viewed as permanent, then scientists conducted a groundbreaking study by activating the MECP2 gene in adult mice that displayed Rett-like symptoms. The experimental results defied predictions because the mice started to heal, their breathing stabilised, mobility returned, and they lived longer [5]. The brain hadn’t forgotten— it had simply been waiting.
Fast forward to today, and gene therapy for Rett has moved from the lab to the clinic. A recent Phase 1/2 trial administered a single dose of AAV-based gene therapy containing a healthy MECP2 gene to girls between 4 to 7 years old who were past the traditional learning window. Doctors documented major improvements in patients after a span of several months. Parents documented new words along with regained motor abilities and independent feeding skills [4,7].
The therapy delivery method stands out because of its exact precision. The MECP2 gene requires precise balance because insufficient levels result in Rett syndrome, but excessive amounts produce a different medical condition [1,2]. The gene therapy uses a shortened MECP2 gene along with a built-in safety switch that keeps protein levels in balance, turning the gene down if too much is made[4,7]. This “Goldilocks” approach— just enough, not too much— highlights how far the science has come.
Beyond Rett: A Broader Future
Rett may be the first neurodevelopmental disorder to show clinical improvement from gene therapy beyond the critical window, but it won’t be the last.
The field of gene therapy now focuses on treating various neurodevelopmental disorders. The University at Buffalo researchers used AAV9 vectors to deliver FOXG1 gene therapy to infant mice with FOXG1 syndrome in 2024, which successfully corrected abnormalities and malformations in different brain regions. The preclinical research provides optimism for upcoming medical interventions that focus on related conditions [5].
Jaguar Gene Therapy launched JAG201 as a Phase 1 clinical trial in 2025 to treat SHANK3 gene disorder, which causes autism spectrum disorder and Phelan-McDermid syndrome. The clinical trial includes both paediatric and adult patients, reflecting growing confidence in the brain’s capacity to respond to gene therapy beyond early development [6].
How It Works: A Genetic Rewrite
Gene therapy functions through the delivery of precise genetic instructions into cells which serve as a replacement for faulty code. The majority of gene therapy methods use AAV (adeno-associated virus) vectors to deliver therapeutic genes into neurons. The gene starts producing the needed protein after entering the cell [5,6,7].
The AAVs function as genetic instruction delivery vehicles. The therapeutic gene moves through body barriers to reach its target cells while delivering the precise genetic message to its intended destination (Figure1).
Scientists continue to develop solutions to overcome the brain’s protective blood-brain barrier, but new solutions are being discovered. Gene therapy in Rett trials involves delivering the treatment through cerebrospinal fluid which enables AAVs to distribute throughout the brain without requiring surgical procedures. The therapy becomes integrated into neurons after delivery and maintains long-term protein production following a single treatment dose.
The recent developments in gene therapy enable the treatment to enter areas it previously could not reach and function where it previously failed to do so.
A New Timeline of Hope
Early intervention is still important. A brain given what it needs from the start has the best chance to thrive. But what science is now revealing, through preclinical breakthroughs and early clinical trials, is that hope doesn’t disappear after the age of two.
For families, this shift is profound. The fear that time is slipping away is being replaced by a renewed sense of possibility.
The critical window may close.
But gene therapy is learning how to open it again.
Bibliography:
- Hensch, T. K. (2004). Critical period regulation. Annual Review of Neuroscience, 27, 549–579. DOI: 10.1146/annurev.neuro.27.070203.144327.
- Daci, R. & Flotte, T. R. (2024). Delivery of adeno-associated virus vectors to the central nervous system for correction of single gene disorders. International Journal of Molecular Sciences, 25(2): 1050. DOI: 10.3390/ijms25021050.
- Creson, T.K., Rojas, C., Hwaun, E., Vaissiere, T., Kilinc, M., Jimenez-Gomez, A., et al. (2019). Re-expression of SynGAP protein in adulthood improves translatable measures of brain function and behavior. eLife, 8, e46752.
- Taysha Gene Therapies, Inc. (2023, June 5). Taysha Gene Therapies announces first patient dosed with TSHA-102 in the Phase 1/2 REVEAL trial under investigation for the treatment of Rett syndrome [Press release]. Retrieved from https://ir.tayshagtx.com/news-releases/news-release-details/taysha-gene-therapies-announces-first-patient-dosed-tsha-102
- Guy, J., Gan, J., Selfridge, J., Cobb, S., & Bird, A. (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science, 315(5815), 1143–1147.
6. Jeon, S., Park, J., Likhite, S., Moon, J.H., Shin, D., Li, L., Meyer, K.C., Lee, J.W., & Lee, S.-K. (2024). The postnatal injection of AAV9-FOXG1 rescues corpus callosum agenesis and other brain deficits in the mouse model of FOXG1 syndrome. Molecular Therapy – Methods & Clinical Development, 32, 101275.
Jaguar Gene Therapy (2024, July 15). Jaguar Gene Therapy to initiate inaugural pediatric clinical trial targeting a genetic form of autism spectrum disorder and Phelan-McDermid syndrome [Press release]. Retrieved from https://www.jaguargenetx.com/newsroom
Related Posts