'Huge congratulations to Jasmine Ardeleanu, runner up of the BSGCT Scientific Writing competition 2026 describing…

BSGCT Scientific Writing Competition – Runner up – Asiya Fatima
‘Huge congratulations to Asiya Fatima, runner up of the BSGCT Scientific Writing competition 2026 describing ‘Why timing decides the fate of babies with SMA’
Asiya Fatima – Why Timing Decides the Fate of Babies with SMA
“I am an MSc graduate in Stem Cells and Regenerative Medicine, with research experience focused on the interface between cellular biology and disease processes. My MSc research investigated the influence of the Parkinson’s protein, α-synuclein on platelet function in healthy volunteers to better understand thrombotic risks in Parkinson’s disease, combining molecular investigation with clinically relevant questions.
Alongside laboratory research, I am passionate about scientific communication as a means of translating complex biomedical concepts into meaningful patient-focused discussions, reflected in being named a runner-up in the BSGCT Scientific Writing Competition 2026. My scientific interests are driven by a commitment to understanding disease mechanisms, improving therapeutic strategies, and contributing to research that bridges bench science with real-world clinical impact. I aspire to grow as a scientist in collaborative research environments where curiosity, precision, and innovation can help advance treatments for complex and currently unmet medical challenges.”
Why Timing Decides the Fate of Babies with SMA
Imagine a race where the outcome is decided before the starting gun is even fired. Now imagine that race involves a newborn baby – one who looks completely healthy in their first moments of life. There is no visible urgency, no sign that anything is wrong. But in some genetic conditions, the race has already begun before birth, unfolding quietly
while everything still appears normal. By the time symptoms appear, the disease is already ahead, and what looks like a starting line is, in reality, a delayed entry.
Spinal muscular atrophy (SMA) makes this reality impossible to ignore.
To understand why, picture how movement works in the body. Motor neurons, which are specialised nerve cells, act like electrical cables, carrying signals from the brain to the muscles. For these cables to stay intact, they depend on a crucial protein called survival motor neuron (SMN) (Fig. 1). This protein functions like a maintenance crew, keeping the communication lines for movement working smoothly.
In SMA, this maintenance system breaks down.
This is due to the gene responsible for producing this protein, SMN1, being faulty or missing (Fig. 1) (1). The body
does have a backup gene, SMN2, but it is inefficient. Most of what it produces is unstable – like spare parts that cannot quite be used (1). Without enough SMN protein, the cables begin to fail. Signals weaken. Muscles gradually lose their connection to the brain (Fig. 1).
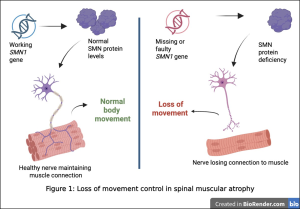
The severity of SMA varies, but its most serious form has painful consequences. In Type I SMA, babies seem healthy at birth, yet within the first few months, something begins to change. Movements that should come naturally, such as lifting the head, rolling over, or sitting, become difficult or never happen at all. While most infants are learning to sit by around six months, babies with SMA are often unable to reach this milestone (2, 3).
Without treatment, many do not survive to see their second birthday (3).
What makes this especially devastating is that the damage does not begin when symptoms appear. It begins earlier,
silently, as motor neurons are progressively lost (4). By the time the condition is recognised, part of the system we are trying to protect is already gone.
For decades, there was little that could be done to change this. Today, that is no longer true.
Gene therapy such as Zolgensma is now transforming the outlook for SMA. By delivering a working copy of the missing SMN1 gene into cells, it restores the body’s ability to produce the SMN protein (3). In doing so, it can
stabilise the remaining neural connections, allowing signals to continue reaching muscles and slowing further damage. But there is a limit. The therapy cannot bring back motor neurons that have already been lost. It can only protect what is still there (3). And that makes timing everything (Fig. 2).

The impact of this timing becomes clear when we look at how children respond to treatment. According to a
preliminary study, babies identified through newborn screening began treatment within the first few weeks of life, at a point where motor neurons were largely preserved. Many of these children were able to achieve developmental
milestones such as sitting, standing, and even walking. In contrast, children diagnosed after symptoms appeared began treatment much later, often after months of progression. Although they received the same therapy, their outcomes were very different. Progress was slower, and recovery was limited (5). What differed was how much of the body’s “wiring” remained by the time treatment was given.
The same therapy, given at a different time, leads to a different life.
This understanding is already reshaping how care is delivered in many parts of the world. Across most of the United States, newborns are routinely screened for SMA at birth (6). Countries including Germany, Belgium, Australia, Japan, and Italy have also introduced SMA into their screening programmes (6). For these children, the race does not begin with visible decline. It begins at a point where the outcome can still be changed.
In the UK, however, the starting point is often later. Newborn screening currently includes only a limited number of genetic conditions, and SMA is not yet widely part of the programme (7). As a result, many children are diagnosed only after symptoms emerge, when valuable time has already been lost. Clinicians have reported stark differences in outcomes compared to countries where early screening is in place (8).
The good news is that there are clear signs of progress. In Scotland, SMA has now been introduced into the newborn screening programme, allowing babies to be tested shortly after birth (9). Across the rest of the UK, further
evaluations are exploring how screening could be expanded, drawing on evidence from countries that have already implemented it (10). The direction is becoming clearer, even if the transition is still in progress.
Beyond treatment itself, timing also shapes everything that follows. Children with SMA often require long-term support, including physiotherapy, respiratory care, and ongoing medical management. For families, the impact is not only clinical but deeply personal. Starting treatment earlier can preserve more function, reducing the level of long-term support (11). In that sense, early diagnosis does more than improving outcomes; it changes the scale of care that follows.
We are making rapid progress in understanding conditions like SMA and developing ways to treat them. But the challenge is no longer only about what we can do; it is about when we do it.
Because in this condition, the race is not won by trying to catch up once symptoms appear.
It is won by recognising that the race has already begun and making sure we are there before the starting gun is even heard.
References:
- Sutherland CS, et The influence of genotype on the natural history of types 1-3 spinal muscular atrophy. Neuromuscul Disord. 2025; 47:105270. DOI: 10.1016/j.nmd.2024.105270
- Cances C, et Natural history of type 1 spinal muscular atrophy: a retrospective, global, multicenter study. Orphanet J Rare Dis. 2022; 17:300. DOI: 10.1186/s13023-022-02455-x
- Canadian Agency for Drugs and Technologies in Zolgensma (Onasemnogene abeparvovec): Clinical review report. Ottawa (ON): Canadian Agency for Drugs and Technologies in Health; 2021
- Glascock J, et Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J Neuromuscul Dis. 2018; 5(2):145-158. DOI: 10.3233/JND-180304
- Krosschell KJ, et Impact of newborn screening and early disease modifying treatment on motor function in spinal muscular atrophy (SMA). Muscular Dystrophy Association (MDA) Clinical & Scientific Conference; 2026. Available from: https://www.mdaconference.org/abstract-library/impact-of-newborn-screening-and-early-disease-modifying-treatment-on-motor-function-in-spinal-muscular-atrophy-sma/
- Cooper K, et Systematic review of newborn screening programmes for spinal muscular atrophy. Int J Neonatal Screen. 2024; 10(3):49. DOI: 10.3390/ijns10030049
- Newborn blood spot screening. [Updated 2024 Sep 5; accessed 2026 Mar 25]. Available from: https://www.nhs.uk/baby/newborn-screening/blood-spot-test/
- Servais L, Dangouloff T, Muntoni F, Scoto M, Baranello Spinal muscular atrophy in the UK: the human toll of slow decisions. The Lancet. 2025; 405(10479):619-620. DOI: 10.1016/S0140-6736(25)00048-0
- Scottish New screening for rare condition in newborns. 2026 [accessed 2026 Mar 25]. Available from: https://www.gov.scot/news/new-screening-for-rare-condition-in-newborns/
- Harcombe Progress made on plans to evaluate SMA newborn screening. UK National Screening Committee; 2025 [accessed 2026 Mar 25]. Available from:
https://nationalscreening.blog.gov.uk/2025/12/04/progress-made-on-plans-to-evaluate-sma-newborn-screening/
- Kariyawasam DS, et Newborn screening for spinal muscular atrophy in Australia: a non-randomised cohort study. Lancet Child Adolesc Health. 2023; 7(3):159-170. DOI: 10.1016/S2352-4642(22)00342-X
Related Posts


